研发中心
AIDD/CADD研发中心
陶术生物AIDD/CADD研发中心专注于计算机辅助药物设计(CADD)和人工智能药物设计(AIDD),为全球药物研发工作者提供从虚拟筛选、分子对接、分子动力学模拟到蛋白设计等全方位技术服务。我们整合了千万级化合物数据库,结合先进的AI算法和超算平台,助力客户加速药物研发进程,攻克研发难题。
专业技术平台 / Professional technical platform

CADD平台
CADD平台提供全方位的计算机辅助药物设计服务,涵盖分子对接、虚拟筛选、分子动力学模拟和反向找靶等。通过分子对接,我们精准预测蛋白与小分子的结合模式;利用虚拟筛选,快速锁定潜在药物分子;结合分子动力学模拟,评估药物稳定性与结合机制。反向找靶技术则帮助客户快速定位小分子的潜在作用靶点,为药物作用机制研究提供关键支持。

AIDD平台
AIDD平台融合前沿AI技术,自主研发了AI筛选方法PLANET和反向找靶算法COMET,提供高效的分子筛选、蛋白设计和反向找靶服务。PLANET基于图神经网络模型,能够从海量化合物库中精准识别潜力分子,显著提升筛选效率,缩短研发周期;COMET结合图神经网络与随机森林技术,快速分析海量靶点数据,精准定位目标分子对应的靶点,极大提升反向找靶效率,加速药物研发进程。

超算平台
超算平台提供强大的高性能计算支持,适用于分子动力学模拟、大规模虚拟筛选和AI模型训练。凭借卓越的算力,我们能够快速处理海量复杂数据,确保科研任务高效完成。平台配备专业的技术团队,提供全方位的技术支持与运维保障,助力客户突破科研瓶颈,加速科研成果转化。

大数据平台
大数据平台整合了TopScience Database千万级小分子数据库和PDBbind生物分子复合物亲合性数据库,为分子对接、虚拟筛选、药-靶作用图谱等研究提供高质量的数据基础。TopScience Database涵盖丰富的化合物信息,支持高效的分子筛选与设计;PDBbind则提供精确的蛋白-配体结合数据,助力药物作用机制研究。两大数据库的结合,为药物研发提供了强大的数据支持,显著提升了研究效率与准确性。
核心团队成员 / Core member
王任小博士,是复旦大学药学院特聘研究员,国家杰出青年。主要研究领域为分子靶向药物设计新方法的发展和应用。代表性成果有对蛋白-配体相互作用打分函数的研究、蛋白-配体复合物数据库、配体分子的自动设计方法等,累计已获得国家发明专利和软件著作权 40 余项,学术生涯已发表 SCI论文 150 余篇(谷歌学术统计他人引用 17000余次,H指数为 51)。 曾先后获得中国化学会“青年计算化学家奖”、中国药学会-施维雅青年药物化学家”、“药明康德生命化学研究奖”等国内知名奖项以及国际化学信息学会“Corwin Hansch Award”。目前兼任中国化学会计算机化学专委会副主任、中国化学会化学生物学专委会委员、中国生物信息学学会药物发现专委会委员、上海市药学会药物化学专委会委员以及美国化学会J.Chem.Inf.Model,等多家国内外学术期刊编委。

王任小博士
陶术生物AIDD/CADD研发中心首席技术官

我们的服务 / Our Services

虚拟筛选
虚拟筛选是一种用于从化学空间中识别潜在活性化合物的技术,传统高通量筛选虽然全面,但识别活性化合物的概率低,且耗费的时间与成本高。而虚拟筛选提高了发现活性化合物的可能性,这使其成为一种更有效和更具成本效益的方法。以美国Structure Bioinformatics Inc. (SBI)提供的数据为例,平均每个新靶点需筛选10万个化合物,其命中率在0.1%~0.01%,而虚拟筛选的命中率可提高到5%~20%,可减少99.9%的费用。

分子对接
分子对接是指受体和配体通过能量匹配、空间匹配、化学性质匹配相互识别形成分子复合物,并预测复合物结构的计算技术。

结构预测
结构预测(同源模建)是基于已知蛋白质结构来预测未知蛋白质结构的方法。它利用蛋白质序列的同源性,即如果两个蛋白质具有足够高的序列相似性,它们很可能具有非常相似的三维结构。当目标蛋白的晶体结构未知时,可以通过寻找与之序列相似的已知结构作为模板,构建模型。

分子动力学模拟
分子动力学是基于经典力学的一种分子模拟方法。与分子力学不同,分子动力学求解的是随时间变化的分子的状态、行为和过程。该方法模拟分子运动的过程,它按照分子瞬时的运动状态,求解每一个原子的牛顿运动方程和每一个原子的位置和速度,并从这一运动轨迹中计算得到各种性质。如果在所研究的模拟时间内能正确地选取分子体系的力场函数形式及其参数,分子动力学就可成为解析分子运动性质的一种强大工具。

反向找靶
反向找靶,也被称为靶点垂钓、反向分子对接等。对于化合物已知疗效,但是,并不知道该化合物产生效果的机制时候,一般会采用反向找靶的方法。潜在药物靶点的识别对于早期药物开发、安全性评估和药物再利用至关重要。然而,由于通量、准确性和成本的限制,实验方法难以广泛应用。作为一种快速、低成本的替代方案,人工智能辅助反向找靶算法的发展越来越受到人们的关注,这些算法可以快速阐明化合物的作用机制。

多肽、蛋白设计
随着对蛋白质功能研究的深入和应用领域的不断拓展,天然蛋白质已难以满足日益增长的需求。因此,蛋白质修饰与设计逐渐转向理性设计甚至从头设计。这种设计范式的转变意味着我们可以超越自然选择的过程,通过对氨基酸序列和三维结构的精确控制来实现特定的蛋白质功能。

PROTAC设计
Protac一般由三部分组成:与靶蛋白结合的配体,用于引导小分子的靶向定位(常选用目标蛋白抑制剂进行衍生);与E3连接酶结合的配体,用于引导蛋白降解;以及负责嵌合两个配体的连接子linker。

先导化合物结构优化
在现代药物研发中,找到一个能够有效治疗疾病的新药是一个复杂且漫长的过程。其中,将苗头化合物(Hit)优化为先导化合物(Lead Compound)是至关重要的一步。这一过程不仅决定了后续开发的方向,也直接影响最终药物的成功与否。
合作案例 / Collaboration case
案例1:助力知名医院医生研究小分子和靶点蛋白结合机制
项目背景及需求
癌症细胞通过代谢重塑适应快速增殖,其中酪氨酸分解代谢产物富马酸对DNA修复机制具有调控作用,影响化疗敏感性,但其具体机制尚不明确。客户推测富马酸可能与富马酸乙酰乙酸水解酶(FAH)结合,进而影响疾病的发生发展,因此需要通过分子模拟方法揭示其结合模式与作用机制,为化疗敏感性研究提供理论依据。
解决方案
AIDD/CADD研发团队建议通过分子对接和分子动力学模拟去分析富马酸和FAH的结合模式及动力学性质,同时分析FAH在疾病中的机制,探究富马酸对FAH的构象影响,从而影响对应的机制。主要流程如下:
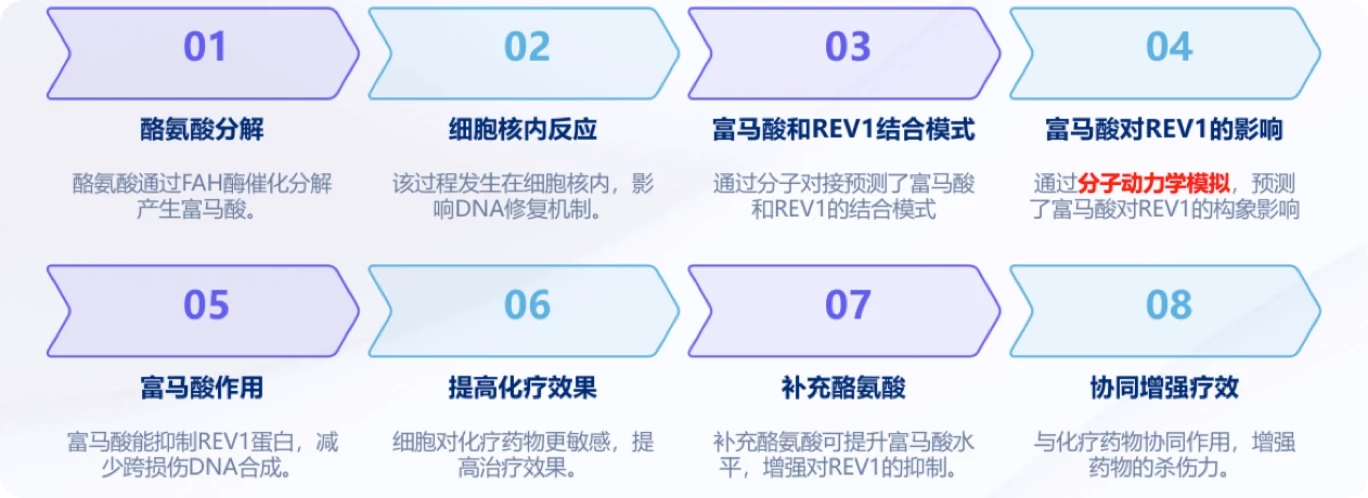
交付成果
本项目提供分子动力学模拟的结果图,展示富马酸对REV1的影响。同时,提供详细的实验流程和细节说明,助力客户文章发表。客户研究成果最后成功发表在《Cell Metabolism》期刊上。

案例2:助力农科院老师发现小麦赤霉病菌药物分子
项目背景及需求
客户发现小麦赤霉病菌上存在一个潜在药物靶点A,针对该靶点进行药物筛选有望开发出治疗小麦赤霉病的有效药物。希望通过虚拟筛选寻找靶点A的活性化合物,并对其结合模式、动力学性质及作用机制进行深入分析,为后续药物研发提供理论支持。(注:因项目尚未发表,文中靶点名称以“A”代替。)
解决方案
AIDD/CADD研发团队建议通过虚拟筛选,快速找到靶点A可能的活性化合物,然后进行活性测试,根据活性测试结果,选择合适的3个分子进行分子动力学实验,针对实验结果进行合理的机制解释。主要流程如下:

交付成果
研发团队为客户提供了针对靶点A的活性化合物列表,并附有详细的实验流程报告及数据解析。在客户撰写文章时,提供对应的实验方法说明。基于筛选结果,客户选择了3个活性化合物进行后续的动力学研究。
客户口碑是
我们的增长源动力
客户评价 / Customer evaluation
陶术生物AIDD/CADD团队非常专业,提供的数据可靠,值得肯定!期待以后继续合作。

中山医学院
潘超云 教授


陶术生物的AIDD/CADD团队提供的结果非常好,数据清晰明了,图片清晰美观,对应结果讨论分析也很到位!

江苏省农科院
曹淑琳 副研究员